If the seizure does not stop in 3 minutes diabetes mellitus weight loss buy discount glucotrol xl 10mg on line, then emergency medical services should be contacted (1) diabetes diet for weight loss cheap 10 mg glucotrol xl with visa. Long-term pharmacotherapy is probably unnecessary diabetes insipidus insulin discount glucotrol xl 10 mg otc, especially for simple febrile seizures diabete 500 glicemia discount glucotrol xl 10mg with amex. Diazepam is given orally using a dose of 1 mg/kg/day in three divided doses when the child is febrile diabetes mellitus fact sheet cheap 10 mg glucotrol xl visa. Other medications that have been used to prevent recurrences are phenobarbital and valproic acid diabetes oral medications powerpoint 10mg glucotrol xl for sale. Although they can prevent 90% of recurrences of febrile seizures diabetes type 2 nih purchase 10mg glucotrol xl mastercard, they are not without significant side effects diabetes mellitus etymology purchase 10mg glucotrol xl free shipping. Phenobarbital has been associated with behavioral problems (hyperactivity) and hypersensitivity reactions. Valproic acid has a risk of developing fatal hepatotoxicity, thrombocytopenia, weight changes, gastrointestinal problems, and pancreatitis. These medications have been considered in those patients who have focal paralysis after a seizure, multiple seizures in a young child, and high parental anxiety despite reassurance (1,4). Phenytoin and carbamazepine have no demonstrated efficacy in preventing febrile seizures. Despite the frightening appearance of the episode, and the parental belief that their child is going to die, simple febrile seizures remain a benign condition with the majority of children having no neurological sequelae. Although the risk of developing another febrile seizure is moderate, the possibility of epilepsy is very small. For this reason, long-term therapy anticonvulsant therapy is not usually recommended, but practitioners should provide reassurance, education of what to do when their child has another febrile seizure, and antipyretic therapy when a fever is present. Why is it important to know this distinction (think of recurrence risk of febrile seizures, development of epilepsy, and work-up) What are three indications for a child who should be hospitalized for overnight observation Although diazepam (Valium) can be used to prevent recurrences when given at the start of a febrile illness, what are its disadvantages Provisional Committee on Quality Improvement, Subcommittee on Febrile Seizures: Practice Parameter: the Neurodiagnostic Evaluation of the Child With a First Simple Febrile Seizure. Committee on Quality Improvement, Subcommittee on Febrile Seizures: Practice Parameter: Long-term Treatment of the Child With Simple Febrile Seizures. It occurs in 2-5% of all children and is the most common reason for convulsions in children less than 5 years of age. Simple seizures are characterized by being less than 15 minutes duration and generalized. Complex febrile seizures are greater than 15 minutes duration, multiple within 24 hours, and focal. One should have a lower threshold for performing tests and hospitalization in cases of complex febrile seizures. Meningitis, encephalitis, Shigella gastroenteritis, medications and toxins, hypoglycemia, electrolyte abnormalities, shaken baby syndrome, accidental head trauma, and epilepsy. Unstable clinical situation, possibility for meningitis, and parents unreliable or unable to cope with the child developing another seizure. One third of children will have at least another febrile seizure with most occurring within one year of the episode. If seizure does not stop within 3 minutes, then emergency medical services should be contacted. He was lying in bed, breathing deeply, difficult to arouse, and could not stand or move his left arm or leg. There was no prior history of trauma, but he was noted to have a small tongue laceration. Deep tendon reflexes are more brisk on the left and a Babinski sign is present on the left as well. The next day, his left-sided weakness and neurologic abnormalities on exam have resolved. After a discussion with his parents, it is decided to discharge him on no anticonvulsant medications. It is concluded that his lethargic episode was due to an unwitnessed seizure with subsequent post-ictal drowsiness. A second nocturnal seizure occurs a year later, and he is started on carbamazepine. He is treated until age 12 when his medication is weaned off and he does well thereafter. A seizure is a sudden, involuntary, stereotypical, repetitive alteration in behavior, including a change in motor activity, in autonomic function, in consciousness, and/or in sensation, which is caused by hypersynchronous discharges from a group of cerebral neurons (1,2). Epilepsy is a condition in which an individual is predisposed to recurrent seizures because of a central nervous system disorder (although about two-thirds have no identifiable cause). Recurrent seizures are the symptomatic expression of underlying brain pathology, not a disease in the usual sense (3). While all people with epilepsy have seizures by definition, not everyone who has a seizure has epilepsy. Four to six percent of all children will have at least one seizure in the first 16 years of life; however, most of these are benign febrile seizures, and the cumulative risk of epilepsy during this time is only about 1-2% (4,5). In other words, less than one third of children who experience a seizure ever develop epilepsy. Over one half of first-time seizures are simple febrile seizures (see the chapter on febrile seizures) and another third are single isolated seizure events or seizures associated with a non-epileptic medical illness. Overall, about half of the lifetime risk of Page - 562 developing epilepsy is realized during the pediatric period, and it is the most common chronic neurologic disorder seen in children. Although other clinical manifestations may also be relevant, such as the neurologic exam, development, etc. Patients with epilepsy usually have a characteristic seizure type although some may have combinations of the following seizure types. More classifications exist, but this chapter will focus on these four basic categories. Partial or focal seizures are often caused by identifiable focal brain lesions which are related to the seizure activity expressed. They may be associated with an aura which is an altered sensation heralding or characterizing partial seizure activity. Any partial seizure can secondarily generalize into a tonic-clonic seizure which is discussed further below. The others may be attributable to some other process and thus, are frequently not diagnosed as seizures. Complex partial seizures also have focal origins but include an impairment of consciousness which implies alteration of functioning in the mesial temporal lobes, orbito frontal lobes, or in more widespread areas of the brain. The impaired consciousness is usually associated with a lack of understanding and memory of the brief event. They may involve more complicated behaviors such as frantic running, uncontrollable laughing, partial undressing, or facial automatisms. Partial complex seizures used to be called temporal lobe seizures because they often originate in the temporal lobes. Partial complex seizures also used to be called psychomotor seizures because they frequently include behavioral symptoms. Generalized seizures typically involve a loss of consciousness and/or a stereotypic motor activity. Absence seizures involve a few seconds of impairment of consciousness with eye blinking or staring which may occur as clustered events. Myoclonic seizures are usually seen in specific epilepsy syndromes and involve quick muscle jerks usually without associated impairment of consciousness. Tonic, clonic, or tonic-clonic seizures involve the abrupt onset of the described muscle activity for several minutes, often followed by post-ictal confusion and fatigue. Atonic seizures involve a sudden loss of postural tone, usually resulting in an abrupt collapse, which may not have any loss of consciousness. Generalized absence seizures are still sometimes called by their old name, petit mal seizures. Generalized tonic clonic seizures are still sometimes called by their old name, grand mal seizures. Partial simple: Previous names include partial elementary seizures, focal motor seizures. Partial complex: Previous names include temporal lobe seizures, psychomotor epilepsy. The history of the event may classically characterize seizure activity, or a patient may present in status epilepticus (see chapter on status epilepticus), in which case the answer to this question is clear. However, often times with non-medical personnel observing what seems to be seizure activity and the emotional anxiety that accompanies it, whether a seizure truly occurred may be unclear. Phenomena which may seem to be generalized seizure-like activity include: syncope, breath-holding spells, panic attacks, psychogenic seizures/pseudoseizures/conversion reaction, gastroesophageal reflux, staring spells, and startle reflexes (infants). In general, syncope tends to be more gradual in onset, may be posturally related, and is without post-event focal neurologic findings or confusion. Seizures, however, are usually associated more with an abrupt onset, secondary injury, and may have post-ictal confusion, headache, incontinence or focal neurologic signs. Syncope may have some brief clonic or myoclonic extremity movements associated with it which can add to the confusion between the two types of events. In general, most true seizure motor activity does not have a reproducible trigger. If a seizure did occur, an important second question to be determined in evaluating a seizure is whether there were acute and reversible provocative causes such as: excessive stimulant medication or stimulant drug abuse, withdrawal from sedative drugs or alcohol, high fever (see chapter on febrile seizures), hypoglycemia, electrolyte imbalance. Answers to this question will play a pivotal role in selecting immediate therapy and determining future prognosis. Although a patient may have more than a single seizure attributed to these problems, these types of seizures would not typically be classified as epileptic. A third question to be answered in the evaluation of what is determined to be a first-time, not acutely reversible seizure, is whether further seizures are expected to occur. This includes an evaluation to determine if the seizure is symptomatic of other pathology and could result in recurrent seizures. These include the following: Page - 563 Vascular etiologies: Arteriovenous malformation Aneurysm, subarachnoid hemorrhage Stroke Venous thrombosis Blood dyscrasias (eg sickle cell anemia) Vasculitides. Those without a known underlying pathology are described as "cryptogenic" (likely an undetectable pathologic explanation) or "idiopathic" (presumed genetic) (1,4). Further consideration is also required to determine whether the seizure may be part of an epileptic syndrome which, by definition, would imply expected recurrent seizure activity without treatment. Cerebral manifestations include increased blood flow, increased oxygen and glucose consumption, and increased carbon dioxide and lactic acid production. If a patient can maintain appropriate oxygenation and ventilation, the increase in cerebral blood flow is usually sufficient to meet the initial increased metabolic requirements of the brain; however, prolonged seizures may result in permanent neuronal injury (2). Salivation may increase secondary to parotid stimulation with masseter muscle contraction. Respirations may cease or be irregular and the patient may have facial cyanosis due to a tonic increase in intrathoracic pressure and impeded venous return associated with maximal muscle group contractions. Failure of adequate ventilation can lead to hypoxia, hypercarbia, and respiratory acidosis. Prolonged skeletal muscle activity can lead to lactic acidosis, rhabdomyolysis, hyperkalemia, and hyperthermia. Postictally (after the seizure event), effects of the massive neuronal depolarization and metabolic activity may include confusion, lethargy or a comatose state. Vomiting may occur, and patients with impaired consciousness may be unable to protect their airway and are at risk for aspiration. Impaired consciousness may also be associated with airway obstruction from the tongue or respiratory secretions. Head trauma may have precipitated a seizure event, but traumatic falls may also occur interictally and contribute to postictal altered mental status and other injuries. The mechanism is not well understood, but it may be attributed to neuronal dysfunction or neurotransmitter exhaustion. The duration and severity of the seizure do not correlate with the degree of postictal paralysis, and the paralysis is usually, but not always, noted in the area of the focal seizure activity (6). Systemically in the postictal state, deep respirations may be present to compensate for respiratory and metabolic acidosis, and blood pressure and temperature quickly return to normal. Due to the catecholamine surge noted above, patients are usually mildly hyperglycemic. Headache and muscle soreness may also occur in association with muscle fatigue and acidosis. The diagnosis of epileptic seizures involves determining: 1) if seizures occurred, 2) the type of seizures, 3) the cause of the seizures, and 4) if they are characteristic of an epileptic syndrome. Underlying seizure disorder, history of previous seizures or other neurologic disorder Other signs of systemic illness or reasons for provocative causes: headache, vomiting, diarrhea, ataxia, altered mental status. Evolution, motor activity of head, eyes, face, trunk, extremities, other complicating factors (cyanosis, trauma, emesis). Postictal state: Incontinence, confusion/sleepy, headache, focal neurologic deficits, time to recovery of normal function (nearly immediate for syncope, minutes to hours for postictal, but usually less than 24hours) Family history: Seizures, epilepsy, neurocutaneous syndromes, other neurologic disorders Neurologic evaluation should include: time to recovery, retrograde amnesia, speech difficulty, cranial nerves function, herniation signs, posturing, postictal deficits such as Todd paralysis, sensory loss, pathological reflexes, coordination or gait changes Diagnostic tests for seizures are usually low-yield without historical or exam findings to suggest possible abnormalities. Routine screening labs, depending on the setting, may include electrolytes, glucose, Ca and Mg. Hyponatremia and hypoglycemia can cause seizures, whereas hypocalcemia and magnesium abnormalities resulting in hypocalcemia may cause tetany which resembles seizures. Numerous channels are recorded simultaneously from standard electrode placements to map brain electrical activity. Potentially provocative maneuvers (procedures known to provoke seizure potentials) known as activation procedures, such as hyperventilation, photic stimulation. Generalized spiking is usually large and obvious, while focal spikes (especially temporal lobe spiking) may be smaller and more subtle to see. Other generalized patterns may also be definitive such as the 3-per-second spike and slow waves of childhood absence epilepsy (petit mal). Other mixtures of signals may also display characteristically defined patterns such as the mixture of spikes and slow waves that are different in each hemisphere described as hypsarrhythmia which is typical of infantile spasms. Partial seizures with secondary generalization demonstrates focal spikes progressing to generalized spiking. Generalized absence seizures display a 3 per second spike and slow wave pattern which is often precipitated by hyperventilation. Generalized tonic-clonic seizures display generalized spiking (photic stimulation may be a useful activation procedure). Infantile spasms, sometimes seen in severe developmental brain anomalies and tuberous sclerosis, display a hypsarrhythmia pattern (disorganized mixture of spikes and slow waves, different in each hemisphere). Benign epilepsy of childhood (Rolandic seizures) displays centrotemporal spikes or sharp waves ("Rolandic discharges") against a normal background. The Lennox-Gastaut syndrome displays slow spike and waves on an abnormal slow background. Therapy for the acutely seizing patient is described in the chapter on status epilepticus. Short-term anti-seizure medication is used as needed, but no long-term anticonvulsant medication is typically employed. The risk for a second seizure in five-years is approximately 30% whereas it is approximately 46-73% for a seizure with any one of the above risk factors (7). It is not beneficial for children to take daily medication for years to prevent an incident that may not be destined to occur during that time period. The benefits of treatment include reducing the risk of recurrent seizures and their potential consequences such as associated injury, effects on self-esteem, and numerous restrictions such as loss of driving license privileges. The patient must be educated about the risk of subsequent seizures and should be advised about state driving regulations (8). Carbamazepine (Tegretol) and phenytoin (Dilantin) are considered the initial medications to consider in all partial seizures and in generalized tonic-clonic seizures (with the exception of infants). Valproic acid (Depakene, Depakote) may be effective both for partial and generalized seizures including absence seizures, but it is typically used only if initial therapy is not successful due to its side-effect profile. The reader is referred to the reference list for further information on these medications and therapy for other epileptic syndromes. The mechanism of action of carbamazepine is thought to be through use-dependent blockade of voltage sensitive sodium channels which results in stabilization of neuronal membranes and inhibition of repetitive firing of neurons. It may be orally or rectally absorbed, has a half-life of 12 to 17 hours and is extensively metabolized in the liver via the cytochrome P450 system. Dose-related side effects of carbamazepine include vertigo, ataxia, diplopia, and drowsiness. Approximately 4% of people treated with carbamazepine develop dermatologic reactions including erythematous and pruritic rashes, toxic epidermal necrolysis, erythema multiforme and Stevens-Johnson syndrome. The onset is usually within the first month of treatment but can be delayed up to 6 months. Serious blood dyscrasias, such as aplastic anemia and agranulocytosis have been reported, and although rare, occur at a frequency 5 to 8 times higher than that of the general population (11). Phenytoin is used for the treatment of simple partial, complex partial, and generalized tonic-clonic seizures. The mechanism of action is similar to carbamazepine by use-dependent blockade of voltage-sensitive sodium channels.
In these individuals diabete 63 purchase glucotrol xl 10mg without prescription, aneurysms diabetic peripheral neuropathy glucotrol xl 10 mg amex, arteriovenous malformations diabetic tattoo generic glucotrol xl 10mg with visa, and dissections are common syndrome x type 2 diabetes cheap glucotrol xl 10mg. Hyperextensibility and joint hypermobility are caused by ligamentous laxity (which predisposes to dislocated hips in infants) diabetes test on arm purchase 10mg glucotrol xl fast delivery. Clubfoot diabetes medications for weight loss quality 10mg glucotrol xl, joint effusions diabetes vomiting order glucotrol xl 10 mg otc, and spondylolisthesis (vertebral displacement) may also be present diabetes type 2 risk factors order glucotrol xl 10mg with mastercard. The gastrointestinal tract can be similarly affected; decrease in tensile strength of the bowel walls predisposes to spontaneous rupture. Individuals diagnosed with E-D usually display one or a combination of these different symptoms. The most severe complications of disease result from bowel and vasculature weakness. Hypermobility syndromes represent the mild end of the spectrum described by the Ehlers-Danlos disorders. Vitamin C helps some individuals who are deficient in lysyl hydroxylase (which uses vitamin C as a cofactor in strengthening collagen fibers). Other Connective Tissue disorders: Homocystinuria is an inborn error of methionine metabolism which results in a Marfan-like syndrome. The two disorders are differentiated by the presence of mental retardation in homocystinuria. Stickler syndrome is a constellation of progressive myopia, sensorineural hearing loss and hypomobility associated with distinct facial features. The diagnosis is suspected in neonates with swollen wrists, ankles or knees, and in children with hearing loss and marfanoid characteristics. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects 2nd edition. Location of fracture (femur and radius vs tibia and radius), type of fracture (comminuted mid shaft vs epiphyseal and greenstick). Careful fracture history, identifying weak bones, and targeting physical therapy to strengthen those bones. Any of the following: pectus carinatum (or excavatum sufficiently severe to require surgery), reduced upper to lower segment ratio, positive wrist and thumb signs, scoliosis greater than 20 degrees of curvature, reduced extension of elbows, medial displacement of medial malleolus causing pes planus, protrusio acetabuli. Any three of the following: hyperextensible doughy skin, atrophic scars, joint hypermobility, connective tissue fragility, and bruising. Marfan syndrome, unlike homocystinuria, is not associated with mental retardation. On the phone is a lab technician from another state who reports that a newborn screen reveals a positive test for galactosemia in one of your patients. The child was discharged home from the hospital at just under 48 hours of age, he appeared well with only a 3% weight loss and mild facial jaundice upon follow-up at day 4 of life. His parents reported no breast feeding difficulties and, despite their concern for the number of hours their new baby slept, the new family appeared to be thriving. Genetic testing has received much attention in the press, with interest focusing on the ethics and repercussions of genetic information. Despite this attention, most people do not realize that for the past twenty years, almost all newborns in the United States have been screened for a number of genetic diseases. Almost all newborn screen tests are quantitative tests for the presence or absence of metabolic or endocrine molecules. When the concentration of the tested molecule is greater or less than a level determined by the reference lab, the test is reported as positive. In order to provide 100% sensitivity for disease, the level of positive detection must be adjusted to a point at which specificity may be quite poor. Of these, 54 infants were confirmed to have the disease (positive predictive value 0. Improvements in mass spectroscopy are greatly increasing the number of inborn errors of metabolism that can be efficiently screened in all newborn infants. A single gene must be discovered and sequenced, which when altered, produces a recognizable disease state. For every detectable alteration, a single unique test must be created and performed. On the other hand, some diseases such as Duchenne muscular dystrophy or osteogenesis imperfecta, may result from one of hundreds of different possible alleles. Cystic fibrosis represents a middle ground, in which hundreds of disease alleles exist, but only a handful produce the vast majority of illness in select populations. In European descendants, the delta F508 allele represents 70% of disease alleles in that gene pool and four additional alleles represent another 10-20%. As more disease alleles are discovered, more tests can be run to determine if each allele is present in a given patient. Each matching test involves a small unique section of single stranded nucleic acid which is glued to the wafer in a specific grid position. There are several other methods to detect genetic based illnesses, including the visual inspection of chromosomes, the augmented inspection of chromosomes using fluorescent antibodies, and multiple methods of detecting the presence, absence and relative quantity of proteins. In theory, the information to predict susceptibility to all genetically based disease is available in zygotes and ancient human remains. As science progresses and discovers how genetic information predicts disease states, the ethical debates over how best to use the information must also progress. Page - 129 Since the discovery that genetic information can predict disease states, people have been afraid that this information might be used in a discriminatory manner. One such fear is the possibility that insurance companies might use genetic test results to increase rates or even deny coverage to individuals with genetic susceptibility to expensive illnesses. A panel of lawyers, genetic counselors, and geneticists reported (at the 1999 meeting of the American Society of Human Genetics) that they had been unable to identify any cases of discrimination by health insurers (6). Simply stated, gene therapy is medicine practiced with a nucleic acid based pharmacy. The patient is given nucleic acids in order to modify a pathologic pattern of protein expression. The treatment for refractory leukemia involves massive chemotherapy to destroy all cancerous cells that are then replaced by a population of cells with "normal" cell cycle regulation. The more classical definition of gene therapy requires the modification of protein expression in existing cells. In creating vectors, scientists remodel viruses, retaining the machinery to identify and infect specific cells (adenoviruses which preferentially affect respiratory epithelium, lentiviruses which preferentially attach to T-cells, and herpes viruses which recognize neurons) but change the genetic material which the virus inserts into the cells. Despite the elegance of these theories, no disease state has been "cured" using gene therapy in a human patient as of this writing. The failure of gene therapy to date, is attributable to both vector and nucleic acid design, which are limited by our rudimentary knowledge of basic cell and molecular biology. The dream of the Human Genome Project, that one day patients will provide a drop of blood, a scraping of cheek cells, or a hair follicle and be provided with a set of probabilities of acquiring all disease states and a range of treatment options based on targeted gene therapy, is far from being realized. In 2000, five years ahead of projections, the first working draft sequence of a human genome was completed. True/False: Current newborn screening can diagnose a handful of inborn errors of metabolism like Galactosemia Describe the various methods of introducing nucleic acids into a cell to alter disease states. Sequence knowledge of the disease locus and mutant alleles and the 1:1 correlation of test to disease allele. For disease conditions with multiple mutant alleles, all possibilities must be specifically tested. The disease does not affect the patient until adulthood when she can make her own decisions. There is no effective prophylactic treatment for a child that will prevent the illness before she reaches adulthood. Testing may be appropriate for a 17 year old who desires pregnancy, has the consent of her parents, and who plans to make the decision to become pregnant based on the information of the test. G, the pediatric chief resident, rushes to the delivery room to assist with a resuscitation being attended by a first year resident and a medical student. A chest X-ray demonstrates severe demineralization of all the bones and multiple rib fractures. A skeletal survey demonstrates severe osteopenia and multiple fractures with crumpling of the long bones. On the pediatric floor, there is a teenager with "osteogenesis imperfecta" who has sustained a tibia fracture. Also, why does one case have a negative family history, while the other case has a positive family history A second year resident mentions that this is similar to muscular dystrophy in which some cases are very severe (with no family history) and other cases are milder with a teen or adult onset (and sometimes with a positive family history). The topic of genetic diseases is therefore broad and encompasses both inherited diseases as well as somatic diseases caused by spontaneous mutations. This chapter covers the mechanisms of gene and chromosome mutation and their relevance to both inherited and somatic diseases. Mendelian genetics and chromosome disorders associated with a variety of clinical conditions will also be reviewed. A single somatic cell that mutates will be the progenitor for a clonal population of cells known as the mutant sector. This "patch" of developing mutant cells tends to stay close together and is phenotypically distinct from the surrounding population of normal somatic cells. If the mutation is compatible with cell survival, phenotypic variations can be visualized such as the pigmented lesions seen in McCune-Albright Syndrome. Somatic mutations are often associated with cancer because they can offer growth advantages. Cancer mutations occur in a special category of genes called protooncogenes, many of which regulate cell division. When mutated, such cells enter a state of uncontrolled division, forming a cluster of cells known as a tumor. A germinal mutation occurs in germ cells which are specialized tissue that is set aside during development to form sex cells. If a mutant sex cell participates in fertilization, then the mutation will be passed on to the next generation. It is possible for mosaic germline mutations to occur in which case the mutation can be transmitted to some progeny but not others. Genome mutations involve the loss or gain of whole chromosomes, giving rise to monosomy or trisomy. These mutations are infrequently transmitted to the next generation because they are often incompatible with survival or at least result in reduced fertility. Thus, most monosomies and trisomies are the result of spontaneous events (new mutations). Chromosome mutations result from rearrangement of genetic material and result in structural changes to the chromosome. The most common type of mutations associated with hereditary diseases are gene mutations, the mechanisms of which will be briefly reviewed here. Because these mutations alter the meaning of the genetic code, they are called missense mutations. Sickle cell anemia is a classic example of a coding sequence point mutation which affects the beta-globin chain of hemoglobin. This single amino acid substitution causes the formation of structurally abnormal hemoglobin resulting in a clinically significant hemoglobinopathy. Point mutations can also change amino acid coding sequences into chain terminating stop sequences. Because these new stop sequences do not code for amino acids, they are known as nonsense mutations. An example of this occurrence again involves the betaglobin gene in a severe form of anemia known as beta-thalassemia. Mutations within noncoding sequences can also interfere with protein synthesis at various levels. Point mutations or deletions affecting promoter and enhancer sequences may suppress gene transcription. Lastly, deletions and insertions can cause frame-shift mutations unless the number of base pairs involved is three or a multiple of three. All Mendelian disorders are the result of expressed mutations in single genes with a noticeable phenotypic effect. The number of these disorders is great with some estimates listing more than 5000 disorders. As the name implies, most of these conditions follow classic Mendelian patterns of inheritance. Although gene expression is usually described as dominant or recessive, in some cases, both of the alleles of a gene pair may be fully expressed in the heterozygote, a phenomenon known as codominance. Histocompatibility and blood group antigens are good examples of condominant inheritance. For instance, some individuals inherit the mutant gene but are phenotypically normal. This is referred to as reduced penetrance, a termed that is expressed mathematically as the percentage of patients that phenotypically express their genotypic mutations. On the other hand, variable expressivity occurs when a trait is seen in all individuals carrying a mutant gene, but is expressed differently. Autosomal dominant disorders usually do not involve diseases where there is a loss of function of an enzyme. Because a 50% reduction of most enzymes can be compensated by the 50% that remain viable. Instead, autosomal dominant disorders usually affect non-enzyme proteins that can be divided into two categories. The first involves proteins that are involved in the regulation of complex metabolic pathways that are subject to feedback inhibition. As a consequence of these receptor abnormalities, cholesterol levels are elevated and induce premature atherosclerosis resulting in increased risk of heart disease. The second type of non-enzyme proteins that are affected by autosomal dominant disorders are certain structural proteins. The detrimental effects of reducing levels of a structural protein by 50% become clearer when considering that the abnormal products from a mutant allele can interfere with the assembly of a functionally normal multimeric complex. For example, the collagen molecule is a trimer in which the three collagen chains are arranged in a helical configuration. Each of the three collagen chains in the helix must be normal in order to produce a stable collagen molecule. Even a single mutant collagen chain disrupts the integrity of the trimeric complex. The effects of these autosomal dominant structural protein disorders are seen in conditions such as osteogenesis imperfecta (occult types), Marfan syndrome, and Ehlers-Danlos syndrome. A simplistic way of looking at this is to consider structural protein mutations like a wall of bricks composed of 50% normal bricks and 50% defective bricks (from the abnormal allele). Disease states with an autosomal recessive inheritance pattern comprise the largest category of Mendelian disorders. Because autosomal recessive disorders require that both parents have the mutant allele, such disorders are characterized by the following features: 1) the trait does not usually affect the parents, but siblings may show the disease, and 2) siblings have one chance in four of being affected with a recurrence risk of 25% for each subsequent birth. In contrast to autosomal dominant disorders, the following features generally apply to autosomal recessive disorders: 1) the expression of the defect tends to be more uniform than in autosomal dominant disorders. There are no Y-linked diseases because the only functional gene on the Y chromosome is the determinant for testes. If this gene is mutated, then the person is infertile and hence, no inheritance is possible. In terms of X-linked recessive inheritance, the heterozygous female usually does not express the full phenotypic change because of the normal paired allele on the other X chromosome. However, because of random inactivation of one of the X chromosomes in females (a phenomenon known as Lyonization), there is a remote possibility for the normal allele to be inactivated in most cells, thereby permitting full phenotypic expression. Therefore, an affected male may pass on the abnormal X allele to his daughter, who may also receive an abnormal X allele from her unaffected heterozygous mother. Thus, this is another mechanism that a female could be affected by an Xlinked recessive disorder. This is not possible if the affected condition is incompatible with survival to reproductive age. These disorders are transmitted by an affected heterozygous female to half her sons and half her daughters and by an affected male parent to all his daughters but none of his sons. The aberrations underlying chromosome disorders may take the form of an abnormal number of chromosomes or alterations in the structure of one or more chromosomes. Aneuploidy refers to conditions where errors occur during meiosis or mitosis that result in the formation of cells with a set of chromosomes that are not a haploid multiple. Subsequently, fertilization of such gametes by normal gametes yields a trisomic zygote (2n+1) or a monosomic zygote (2n-1) respectively. In anaphase lag, one homologous chromosome in meiosis or one chromatid in mitosis lags behind, is left out of the cell nucleus and eventually undergoes degeneration. Anaphase lag is similar to nondisjunction except that the chromosome or chromatid gets lost, so that one daughter cell has the right number of chromosomes and one daughter cell has one less than normal. In the former case, fertilization with a normal gamete will form a zygote with one less chromosome yielding a true monosomic zygote. In the latter case, if anaphase lag occurs after the zygote has already formed, a mosaic, composed of normal cells and monosomic cells, is produced. Autosomal monosomy generally involves the loss of too much genetic information to permit live birth or even embryogenesis.
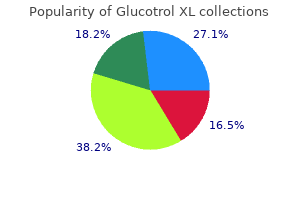
Thus diabetes type 2 zwangerschap buy 10mg glucotrol xl with mastercard, copies of two (2) drug screen results diabetes kit bag discount glucotrol xl 10mg free shipping, one (1) dated within the previous three (3) months must be provided for all renewal requests diabetic diet 55 order glucotrol xl 10 mg free shipping. Medical records/chart notes may be submitted instead of drug screen labs (same timeframe applies) blood sugar guidelines for diabetics cheap 10 mg glucotrol xl with mastercard. The prescriber must sumbit an attestation that the member had consistent participation in a substance abuse or behavioral health treatment program diabetes ketogenic diet discount glucotrol xl 10 mg amex, behavioral health counseling diabetes type 2 list of foods to eat glucotrol xl 10 mg discount, or an addictions recovery program diabetes epidemic cheap glucotrol xl 10 mg with visa. For Suboxone and Subutex diabetes type 1 new york times buy 10mg glucotrol xl mastercard, the prescribing physician must document that the continuation therapy is an attempt at a step-down dose. Fluoroquinolone resistance in ophthalmology and the potential role for newer ophthalmic fluoroquinolones. Cost-effectiveness of linezolid and vancomycin in the treatment of surgical site infections. Bactericidal activity of orally available agents against methicillin-resistant Staphylococcus aureus. Linezolid for the treatment of patients with endocarditis: a systematic review of the published evidence. Approaches to serious methicillin-resistant Staphylococcus aureus infections with decreased susceptibility to vancomycin: clinical significance and options for management. Treating foot infections in diabetic patients: a randomized, multicenter, open -label trial of linezolid versus ampicillin-sulbactam/amoxicillin-clavulanate. Role of linezolid in the treatment of complicated skin and soft tissue infections. Requests for continuing therapy that were approved by a previous Health Plan will be honored for at least 30 days upon receipt of documentation demonstrating that approval. For instance, since the advent of atypical antipsychotics we have seen an increase in use up to 600%; compared to an increase in psychotherapy of 70% during that same time. In these reports, Oklahoma consistently ranks low compared with other states indicating that Oklahoma youth have higher prevalence of mental illness and lower rates of access to care. Therefore, it is imperative that up-to-date evidence-based resources and collaboration is available to our clinicians on the front line of what at times can feel like a mental illness epidemic. It is important to note that the majority of psychotropic medications are prescribed by clinicians with limited training in child and adolescent psychiatry, and Oklahoma is no exception. With the severe shortage of child and adolescent psychiatrists and limited access to evidence-based therapy, clinicians are doing what they can, with the information they know, to treat the symptoms of often devastating and destructive mental health symptoms in our youth. Unfortunately, these interventions are often not-evidence based, masking the underlying disease state rather than treating the underlying problem. This can cause harmful and sometimes lifelong side effects including but not limited to tardive dyskinesia and metabolic syndrome. Judicious use of psychotropic medications is essential in the holistic well-being of the children of Oklahoma. With this need in mind, the Child and Adolescent Psychiatry Division of Oklahoma State University Center for Health Sciences has assembled a team of local experts to create and disseminate Pediatric Psychotropic Medication Resource Guide for Oklahoma youth. Second-Generation Antipsychotic Drug Use Among Medicaid-Enrolled Children: Quality of Care Concerns. United States Government Accountability Office Report to Congressional Requesters. Oklahoma Pediatric Psychotropic Medication Task Force Clinicians from Oklahoma State University Center for Health Sciences and University of Oklahoma Center for Health Sciences lead the core team. The core team invited task force members to participate in the drafting of the resource guide. Our task force consisted of child and adolescent psychiatrists, pediatricians and pharmacists who reviewed and compiled up-to-date information on best prescribing practices. Task force members were identified through their community standing and clinical expertise. Task force members were responsible for reviewing current research practices along with thoughtful clinical acumen to prepare the specific topics included in the resource guide. Through collaboration and consensus building, a first draft of the Oklahoma Pediatric Psychotropic Medication Resource Guide was developed. The details in this report rely on the most up-to-date evidence through December 2019. Subsequent revisions in the coming years will be made available to ensure our treatment recommendations are evidence-based and current. Although this resource is meant to aid in the diagnosis and treatment of children and adolescents, it is important to note that ultimately, the clinical decision making relies on the treating clinician and treatment team. Best efforts should be made to obtain all past medical history for outpatient and inpatient treatment. Each clinician should determine their comfort diagnosing and treating based on their training and expertise. When indicated, clinicians should seek further consultation through available methods. For the majority of psychiatric diagnosis, behavioral therapy (including caregiver participation) is indicated as first-line treatment. Rating scales for disorders should be used as screeners and to ensure treatment response. Typically monitoring of mental health concerns should occur every week to two weeks at first, then monthly as symptoms stabilize to less frequent visits. The assent and consent discussion should focus on the risks and benefits of the proposed and alternative treatments. If multiple medications are indicated, only one medication change should be made at a time unless clinically indicated. Other than cross-tapering,* there is no evidence to support the use of two medications from the same class being used simultaneously and should be avoided. Discontinuing Medications If the patient has shown a sustained period of remission or recovery and the prescriber believes the medication may no longer be necessary, a discontinuation trial may be clinically indicated. Before initiating a discontinuation trial, the plan for discontinuation is reviewed with the patient and family focusing on the risks of discontinuation. This is especially important if the patient was significantly impaired or suicidal before medication treatment. A specific plan for tapering and discontinuing medication and appropriate frequency of monitoring visits prevents withdrawal effects of medication and allows the clinician to identify early relapse or recurrence of symptoms. Monitoring children for a period of time after they are off medication allows for early identification of relapse or recurrence before symptoms become too severe. During the discontinuation phase, patients may need to be seen more frequently than during the maintenance phase. Close monitoring as the dose of medication is being lowered, and for a period of time thereafter, ensures withdrawal symptoms and early signs of relapse or recurrence are identified quickly. At this time, there are little or no data to suggest which medication to remove first in children who are taking multiple medications. Note: When switching psychotropics, medication overlaps and cross taper should occur in a timely fashion, generally within four weeks. Psychotropic Medication Utilization Parameters for Children and Youth in Foster Care (2016). Therefore we highly encourage consultation with a child and adolescent psychiatrist prior to initiating medication. Cool Little Kids2 Opinion/Clinical Opinion Comments Strong Recommendation 2nd-Line Treatment Strong Recommendation M E D I C I N E. Proceedings of the Annual Meeting of the Anxiety Disorders Association of America. The University of South Florida, Florida Medicaid Drug Therapy Management Program sponsored by the Florida Agency for Health Care Administration. There are relationship questions and environmental safety questions below that the clinician can use for decisions about referrals. These should be combined with observations made in the clinic of relationship concerns between the child and the parent. E D U 13 Behavioral screening (emotion, conduct, hyperactivity, peer problems, prosocial behavior) for children over the age of two. In this age group there are many differential diagnoses and co-morbidities to assess. As such, therapy should be first line to avoid potential side effects of psychotropic medications. General symptoms screenings can be used to determine the need for a referral for further evaluation. O K L A H O M A S T A T E U N I V E R S I T Y C E N T E R F O R H E A L T H S C I E N C E S 20 Disruptive Behavior Disorders in Young Children. Includes disruptive behavior questions Includes the Baby Symptom Checklist for ages two months to 18 months and the Preschool Pediatric Symptoms Checklist for ages 18-60 months. O K L A H O M A S T A T E U N I V E R S I T Y C E N T E R F O R H E A L T H S C I E N C E S 26 4. Co-morbid conditions should be treated as well, and these conditions may have supporting evidence for psychotropic use. It is recommended that the developmental, emotional/behavioral symptoms and family/ environmental context are all screened. In addition, if there is concern, the young child should be referred to an infant mental health provider. Survey of Well-Being of Young Children Screens three domains-developmental, emotional/behavioral, and family context, including safety questions. O K L A H O M A S T A T E U N I V E R S I T Y C E N T E R F O R H E A L T H S C I E N C E S 28. Appropriate assessment should be conducted prior to treating the symptom of aggression. Physical conditions which may mimic anxiety include hyperthyroidism, caffeinism, migraines, asthma, seizure disorders and lead intoxication. Stage 2A: Parent and child education about anxiety disorders (see resource for Child Mind Institute below). Black Box Warning: children and adolescents have an increased risk of suicidal ideations at therapy initiation and patients should be monitored closely. Tricyclics are no longer recommended because of cardiac monitoring requirements and a greater risk for overdose. If concerns for traumatic stress exist, evidence-based therapy to address the trauma should occur. American Academy of Pediatrics (2019) Subcommittee on Attention-Deficit/Hyperactivity Disorder, Steering Committee on Quality Improvement and Management. American Academy of Child and Adolescent Psychiatry (2007) Practice Parameter for the Assessment and Treatment of Children and Adolescents With Attention-Deficit/Hyperactivity Disorder. O K L A H O M A S T A T E U N I V E R S I T Y C E N T E R F O R H E A L T H S C I E N C E S 44 4. Children who show pragmatic or social language delays should also be referred to speech and language therapy. Life Skills: Daily life skills can be taught by occupational therapists and other providers. Children with autism can be treated with psychotropic medications when there is a specific target symptom or co-occurring behavioral health condition. If irritability is related to an underlying behavioral health issue such as anxiety or depression, treat that accordingly. They should be used cautiously with careful monitoring as potential side effects include sedation, weight gain, cholesterol abnormalities and movement disorders. Consider referral for behavioral therapy or parent child interaction therapy for younger children. Make sure the child has a functional means of communication and refer to a speech and language therapist if warranted. Behavioral activation is a significant concern and lower doses should be considered with slow titration. Antipsychotics are typically used, but only studied in children with psychosis without autism. Refer to a specialist if there is a concern a child with autism may have psychotic symptoms. O K L A H O M A S T A T E U N I V E R S I T Y C E N T E R F O R H E A L T H S C I E N C E S 50 10. ExtendedRelease Guanfacine for Hyperactivity in Children With Autism Spectrum Disorder. Chapel Hill: the University of North Carolina, Frank Porter Graham Child Development Institute, Autism Evidence-Based Practice Review Group. These symptoms are also often in combination with attention deficit hyperactivity disorder and/or disruptive behavior disorders. Type I is when the manic episode lasts at least seven days and/or the child requires hospitalization. Pediatric patients may also experience bipolar depression which may guide medications. Atypical antipsychotics improve manic symptoms significantly more than mood stabilizers in youths7,8 and should be selected first assuming no allergies or contraindications. Risperidone,10,11 aripiprazole,12 and lithium13 help as monotherapy for bipolar mania. Aripiprazole and lithium are not statistically significantly different in treatment of mania symptoms at 12 weeks and both are better than placebo, although aripiprazole may confer higher rates of gastrointestinal disturbances. If there is a partial response to a single agent listed in Stage 2, augment with a medication from another class. Additional options to those not listed in Stage 2 are mood stabilizers valproate and lamotrigine or atypical antipsychotic ziprasidone. If monotherapy with an atypical listed in Stage 2 is ineffective, either (1) switch to a different atypical antipsychotic, or (2) switch to a mood stabilizer. If augmented therapy in Stage 3 is ineffective, ensure therapy is optimized before switching to a new agent in either class. If monotherapy listed in Stage 3 is ineffective, consider dual therapy with a combination of atypical antipsychotic and mood stabilizer. If bipolar depression is of concern, combination olanzapine/fluoxetine19 or lurasidone20 monotherapy may be considered. Other switching strategies may be employed if the clinician determines this method is suboptimal. Please refer to general atypical antipsychotics information found on page 63 for guidance. More research is needed before these agents should be started as treatment for bipolar disorder. O K L A H O M A S T A T E U N I V E R S I T Y C E N T E R F O R H E A L T H S C I E N C E S 56 7. It is important to note that auditory hallucinations alone do not substantiate the diagnosis of schizophrenia. The diagnosis should be made with detailed input of family, teachers, pediatricians, family physicians, etc. Finally, the diagnosis and initial management needs to be made by an adult or child psychiatrist experienced in the evaluation and treatment of adolescents and children. However, if there is no response after two weeks at a therapeutic dose, consider changing to a different agent.
Behavioral control of breathing allows it to be integrated with swallowing diabetes mellitus type 2 histology discount glucotrol xl 10 mg visa, and in humans diabetic neuropathy symptoms purchase glucotrol xl 10 mg amex, with verbal and Cortex emotional communication as well as other behaviors diabetes type 1 zorgverzekering buy glucotrol xl 10mg low cost. This rhythm is regulated in the intact brain by a number of influences that enter via the vagus and glossopharyngeal nerves managing type 1 diabetes in adults discount glucotrol xl 10mg on line. These control airway and respiratory reflexes blood glucose 246 glucotrol xl 10mg, analogous to the cardiovascular system diabetic living recipes buy generic glucotrol xl 10mg, by inputs to the ventrolateral medulla diabetes in spanish generic 10 mg glucotrol xl with amex. These include outputs to the airways via the vagus nerve (red) and outputs from the ventral respiratory group (orange) to the spinal cord diabetes test gp discount glucotrol xl 10mg line, controlling sympathetic airway responses (green) and respiratory motor (phrenic motor nucleus, blue) and accessory motor (hypoglossal and intercostal, blue) outputs. However, it is assisted in this process by the parabrachial nucleus (or pontine respiratory group, purple), which receives ascending respiratory afferents and integrates them with other brainstem reflexes. The prefrontal cortex (brown) provides behavioral regulation of breathing, producing a continual breathing rhythm even in the absence of metabolic need. This influences the hypothalamus (light green), which may vary respiratory pattern in coordination with behavior or emotion. Examination of the Comatose Patient 49 the carotid sinus branch of the glossopharyngeal nerve brings afferents that carry information about blood oxygen and carbon dioxide content, whereas the vagus nerve conveys pulmonary stretch afferents. These terminate in the commissural, ventrolateral, intermediate, and interstitial components of the nucleus of the solitary tract. These influences are relayed to reticular areas in the ventrolateral medulla that regulate the onset of inspiration and expiration. On the other hand, neurons located more ventrally in the intertrigeminal zone, between the principal sensory and motor trigeminal nuclei, produce apneas, which are necessary during swallowing and in response to noxious chemical irritation of the airway. Respiration can be altered by emotional response, and it increases in anticipation of metabolic demand during voluntary exercise, even if the muscle that is to be contracted has been paralyzed. The pathways that control vocalization in humans appear to originate in the frontal opercular cortex, which provides premotor and motor integration of orofacial motor actions. However, there is also a prefrontal contribution to the maintenance of respiratory rhythm, even in the absence of metabolic demand (the basis for posthyperventilation apnea, described below). By contrast, subjects with diffuse metabolic impairment of the forebrain, or bilateral structural damage to the frontal lobes, commonly demonstrate posthyperventilation apnea. Rhythmic breathing returns when endogenous carbon dioxide production raises the arterial level back to normal. The demonstration of posthyperventilation apnea requires that the patient voluntarily take several deep breaths, so that it is useful in differential diagnosis of lethargic or confused patients, but not in cases of stupor or coma. If the lungs function well, the maneuver usually lowers the arterial carbon dioxide by 8 to 14 torr. At the end of the deep breathing, wakeful patients without brain damage show little or no apnea (less than 10 seconds). However, in patients with forebrain impairment, the period of apnea may last from 12 to 30 seconds. The neural substrate that produces a continuous breathing pattern even in the absence of metabolic need is believed to include the same frontal pathways that regulate behavioral alterations of breathing patterns, as the continuous breathing pattern disappears with sleep, bilateral frontal lobe damage, or diffuse metabolic impairment of the hemispheres. Different abnormal respiratory patterns are associated with pathologic lesions (shaded areas) at various levels of the brain. This rhythmic alternation in Cheyne-Stokes respiration results from the interplay of normal brainstem respiratory reflexes. There is normally a short delay of a few seconds, representing the transit time for fresh blood from the lungs to reach the left heart and then the chemoreceptors in the carotid artery and the brain. By the time the brain begins increasing the rate and depth of respiration, the alveolar carbon dioxide has reached even higher levels, and so there is a gradual ramping up of respiration as the brain sees a rising level of carbon dioxide, despite its additional efforts. By the time the brain begins to see a fall in carbon dioxide tension, the levels in the alveoli may be quite low. When blood containing this low level of carbon dioxide reaches the brain, respiration slows or may even cease, thus setting off another cycle. Hence, the periodic cycling is due to the delay (hys- Examination of the Comatose Patient 51 teresis) in the feedback loop between alveolar ventilation and brain chemoreceptor sensory responses. The Cheyne-Stokes respiratory cycle is not usually seen in normal individuals because the circulatory delay between a change in alveolar blood gases and carbon dioxide tension in the brain is only a few seconds. Even as circulatory delay rises with cardiovascular or pulmonary disease, during waking the descending pathways that prevent posthyperventilation apnea also ensure the persistence of respiration even during periods of low metabolic need, thus damping the oscillations that produce CheyneStokes respiration. However, during sleep or with bilateral forebrain impairment, due either to a diffuse metabolic process such as uremia, hepatic failure, or bilateral damage such as cerebral infarcts or a forebrain mass lesion with diencephalic displacement, periodic breathing may emerge. Thus, Cheyne-Stokes respiration is mainly useful as a sign of intact brainstem respiratory reflexes in the patients with forebrain impairment, but cannot be interpreted in the presence of significant congestive heart failure. Some patients hyperventilate when intrinsic brainstem injury or subarachnoid hemorrhage or seizures cause neurogenic pulmonary edema. The pulmonary congestion lowers both the arterial carbon dioxide and the oxygen tension. Stimulation of pulmonary stretch re- ceptors is apparently sufficient to cause reflex hyperpnea, as oxygen therapy sufficient to raise the arterial oxygen level does not always correct the overbreathing. Another small group of patients has been identified who have hyperventilation associated with brainstem gliomas or lymphomas. It is theoretically possible for an irritative lesion in the region of the parabrachial nucleus or other respiratory centers to produce hyperpnea. The respiratory changes must persist during sleep to eliminate psychogenic hyperventilation, and one must exclude the presence of stimulating drugs, such as salicylates, or disorders that stimulate respiration, such as hepatic failure or underlying systemic infection. Cases fulfilling all of these criteria have rarely been observed,50,51 and none that we are aware of has come to postmortem examination of the brain. Fully developed apneustic breathing, with each cycle including an inspiratory pause, is rare in humans, but of considerable localizing value. Clinically, end-inspiratory pauses of 2 to 3 seconds usually alternate with end-expiratory pauses, and both are most frequently encountered in the setting of pontine infarction due to basilar artery occlusion. However, apneustic breathing may rarely be observed in metabolic encephalopathies, including hypoglycemia, anoxia, or meningitis. At least one patient with apneusis due to a brainstem infarct responded to buspirone, a serotonin 1A receptor agonist. The resulting irregular, gasping breathing is eerily similar to humans with bilateral rostral medullary lesions, and it indicates that sufficient neurons survive in the medullary reticular formation to drive primitive ventilatory efforts, despite the loss of the neurons that cause smooth to-and-fro respiration. A variety of intermediate types of breathing patterns are also seen with high medullary lesions. Some patients may breathe in irregular clusters or ratchet-like breaths separated by pauses. In other cases, particularly during intoxication with opiates or sedative drugs, the breathing may slow and decline in depth gradually until it fades into complete arrest. There is a tendency in modern hospitals to intubate and ventilate patients with structural coma to protect the airway and permit treatment of respiratory failure. If the patient fights intubation or ventilation, paralytic drugs are often administered. This compromises the ability of the neurologist to assess brainstem reflexes, and in some cases may delay diagnosis and compromise care. Thus, it is important, whenever possible, to delay intubation until after the brief coma examination described here has been completed. This results in critical narrowing of the airway and the increased rate of movement of air tends to further reduce airway pressure, resulting in sudden closure. Liable to the disorder are obese patients, because deposition of fat in neck tissue reduces airway diameter; men, because the increased ratio of the length of the airway to its diameter predisposes to collapse; and middle aged or older patients, because muscle tone is more reduced during sleep with age. Sleep apnea typically occurs in cycles lasting a few minutes each when the patient falls asleep, airway tone fails and an obstructive apnea occurs, blood oxygen levels fall, carbon dioxide rises, and the patient is aroused sufficiently to resume breathing. The fragmentation of sleep and intermittent hypoxia result in chronic daytime sleepiness and impairment of cognitive function, particularly vigilance. Excessive drowsiness during the day and loud snoring at night may be the only clues. Lethargy or drowsiness due to neurologic injury may induce apneic cycles in a patient with obstructive sleep apnea. However, as the level of consciousness becomes more impaired, it may be difficult to achieve the periodic arousals necessary to resume breathing. Most such patients have congestive heart failure, and the pauses are thought to be analogous to the periodic breathing that is seen in patients who develop Cheyne-Stokes respiration when they fall asleep. Yawning may improve the compliance of the lungs and chest wall, but its function is not understood. It may be seen in lethargic patients, but yawning is also seen in complex partial seizures emanating from the medial temporal lobe, and is not of great localizing value. Because stuporous patients with intracranial mass lesions are often treated with corticosteroids to reduce brain edema, it may be difficult to determine whether pressure on the floor of the fourth ventricle from the mass lesion or the treatment with corticosteroids is causing the hiccups. As an example, one patient in New York Hospital with a low brainstem infarct and tracheostomy maintained his total ventilation for several days by hiccup alone. Agents used to treat hiccups include phenothiazines, calcium channel blockers, baclofen, and anticonvulsants, gabapentin being the most recent. The vomiting reflex may be triggered by vagal afferents75,76 or by chemosensory neurons in the area postrema, a small group of nerve cells that sits atop the nucleus of the solitary tract in the floor of the fourth ventricle, just at the level of the obex. It occasionally occurs in patients with irritative lesions limited to the region of the nucleus of the solitary tract. More commonly, however, vomiting is due to a sudden increase in intracranial pressure, such as occurs in subarachnoid hemorrhage. The pressure wave may stimulate the emetic response directly by pressure on the floor of the fourth ventricle, resulting in sudden, ``projectile' vomiting, without warning. This type of vomiting is particularly common in children with posterior fossa tumors. It is also seen in adults with brain tumor, who hypoventilate during sleep, resulting in cerebral vasodilation. The small increase in intravascular blood volume, in a patient whose intracranial pressure is already elevated, may cause a sharp increase in intracranial pressure (see Chapter 3), resulting in onset of an intense headache that may waken the patient, followed shortly thereafter by sudden projectile vomiting. Vomiting is also commonly seen in patients with brain tumors during chemotherapy or even radiation therapy. The anatomy of these pathways is closely intertwined with the components of the ascending arousal system. In addition, the pupillary pathways are among the most resistant to metabolic insult. Hence, abnormalities of pupillary responses are of great localizing value in diagnosing the cause of stupor and coma, and the pupillary light reflex is the single most important physical sign in differentiating metabolic from structural coma. Examine the Pupils and Their Responses If possible, inquire if the patient has suffered eye disease or uses eyedrops. Observe the pupils in ambient light; if room lights are bright and pupils are small, dimming the light may make it easier to see the pupillary responses. They should be equal in size and about the same size as those of normal individuals in the same light (8% to 18% of normal individuals have anisocoria greater than 0. Unequal pupils can result from sympathetic paralysis making the pupil smaller or parasympathetic paralysis making the pupil larger. Unless there is specific damage to the pupillary system, pupils of stuporous or comatose patients are usually smaller than normal pupils in awake subjects. The eyelids can be held open while the light from a bright flashlight illuminates each pupil. Shining the light into one pupil should cause both pupils to react briskly and equally. Because the pupils are often small in stuporous or comatose patients and the light reflex may be through a small range, one may want to view the pupil through the bright light of an ophthalmoscope using a plus 20 lens or through the lens of an otoscope. Most pupillary responses are brisk, but a tonic pupil may react slowly, so the light should illuminate the eye for at least 10 seconds. Moving the light from one eye to the other may result in constriction of both pupils when the light is shined into the first eye, but paradoxically pupillary dilation when the light is shined in the other eye. In a comatose patient, this usually indicates oculomotor nerve compromise either by a posterior communicating artery aneurysm or by temporal lobe herniation (see oculomotor responses, page 60). However, the same finding can be mimicked by unilateral instillation of atropinelike eye drops. Occasionally this happens by accident, as when a patient who is using a scopolamine patch to avert motion sickness inadvertently gets some scopolamine onto a finger when handling the patch, and then rubs the eye; however, it is also seen in cases of factitious presentation. Still other times, unilateral pupillary dilation may occur in the setting of ciliary ganglion dysfunction from head or facial trauma. In most of these cases there is a fracture in the posterior floor of the orbit that interrupts the fibers of the inferior division of the oculomotor nerve. The denervated pupil will respond briskly, whereas the one that is blocked by atropine will not. A normal ciliospinal response ensures integrity of these circuits from the lower brainstem to the spinal cord, thus usually placing the lesion in the rostral pons or higher. Pathophysiology of Pupillary Responses: Peripheral Anatomy of the Pupillomotor System the pupil is a hole in the iris; thus, change in pupillary diameter occurs when the iris contracts or expands. The pupillodilator muscle is a set of radially oriented muscle fibers, running from the edge of the pupil to the limbus (outer edge) of the iris. When these muscles contract, they open the pupil in much the way a drawstring pulls up a curtain. The pupillodilator muscles are innervated by sympathetic ganglion cells in the superior cervical ganglion. These axons pass along the internal carotid artery, joining the ophthalmic division of the trigeminal nerve in the cavernous sinus and accompanying it through the superior orbital fissure, into the orbit. Sympathetic input to the lid retractor muscle takes a similar course, but sympathetic fibers from the superior cervical ganglion that control facial sweating travel along the external carotid artery. The sympathetic preganglionic neurons for pupillary control are found in the intermediolateral column of the first three thoracic segments. Two summary drawings indicating the (A) parasympathetic pupilloconstrictor pathways and (B) sympathetic pupillodilator pathways. The parasympathetic neurons that supply the pupilloconstrictor muscle are located in the ciliary ganglion and in episcleral ganglion cells within the orbit. The preganglionic neurons for pupilloconstriction are located in the oculomotor complex in the brainstem (Edinger-Westphal nucleus) and they arrive in the orbit via the oculomotor or third cranial nerve. The pupilloconstrictor fibers travel in the dorsomedial quadrant of the third nerve, where they are vulnerable to compression by a number of causes (Chapter 3), often before there is clear impairment of the third nerve extraocular muscles. As a result, unilateral loss of pupilloconstrictor tone is of great diagnostic importance in patients with stupor or coma caused by supratentorial mass lesions. Pharmacology of the Peripheral Pupillomotor System Because the state of the pupils is of such importance in the diagnosis of patients with coma, it is sometimes necessary to explore the origin of aberrant responses. Knowledge of the pharmacology of the pupillomotor system is essential to properly interpret the findings. The sympathetic terminals onto the pupillodilator muscle in the iris are noradrenergic, and they dilate the pupil via a beta-1 adrenergic receptor. In the presence of a unilateral small pupil, it is possible to determine whether the cause is due to failure of the sympathetic ganglion cells or is preganglionic. The pupil can then be dilated by instilling a few drops of 1% hydroxyamphetamine into the eye, which releases norepinephrine from surviving sympathetic terminals. Because the postsynaptic receptors have become hypersensitive due to the paucity of neurotransmitter being released, there is brisk pupillodilation after instilling the eye drops. Conversely, if the pupil is small due to loss of postganglionic neurons or receptor blockade, hydroxyam- phetamine will have little if any effect. Denervated receptors are hypersensitive and there is brisk pupillary dilation, but a pupil that is small due to a beta blocker does not respond. The parasympathetic ganglion cells, by contrast, activate the pupilloconstrictor muscle via a muscarinic cholinergic synapse. In the presence of a dilated pupil due to an injury to the third nerve or the postganglionic neurons, the hypersensitive receptors will constrict the pupil rapidly in response to a dilute solution of the muscarinic agonist pilocarpine (0. However, if the enlarged pupil is due to atropine, even much stronger solutions of pilocarpine (up to 1. Preganglionic sympathetic neurons in the C8-T2 levels of the spinal cord, which regulate pupillodilation, receive inputs from several levels of the brain. The main input driving sympathetic pupillary tone derives from the ipsilateral hypothalamus. Neurons in the paraventricular and arcuate nuclei and in the lateral hypothalamus all innervate the upper thoracic sympathetic preganglionic neurons. Thus, the sympathoexcitatory pathway remains ipsilateral from the hypothalamus all the way to the spinal cord. Brainstem sympathoexcitatory neurons can cause pupillodilation in response to painful stimuli (the ciliospinal reflex). As a result, lesions of the pontine tegmentum, which destroy both these ascending inhibitory inputs to the pupilloconstrictor system and the descending excitatory inputs to the pupillodilator system, cause the most severely constricted pupils seen in humans. Preganglionic parasympathetic neurons are located in the Edinger-Westphal nucleus in primates. In rodents and cats, most of the pupilloconstrictor neurons are located outside the Edinger-Westphal nucleus, and the nucleus itself mainly consists of the spinally projecting population, so that extrapolation from nonprimate species (where the anatomy and physiology of the system has been most carefully studied) is difficult.